Q1:
I am using larva software to investigate the noncoding hotspot mutation, but one error message was reported as follows:
Error: Mutation counts file example.snv.bed has too few columns on line 1. Expected at least 5, but found 4. Exiting.
The command I used: ./larva -vf example.snv.bed -af example.anno.bed -o larva.out -b
It makes me pretty confused that the “example.snv.bed” file really has 5 columns seperated by tab but the error says only found 4. I have tried a lot but still could not figure it out. Could you please give some help?
#####
The example.snv.bed file likes this:
chrM 5650 5651 BLCA_GD blca01
chrM 8863 8864 BLCA_GD blca01
chr1 1111476 1111477 BLCA_GD blca01
chr1 1632977 1632978 BLCA_GD blca01
chr1 1657153 1657154 BLCA_GD blca01
chr1 2584370 2584371 BLCA_GD blca01
####
The example.anno.bed file likes this:
the fourth column is the annotation info(only subset )
It would be really a great appreciate for your help.
A1:
It looks like the variant file and annotation file excerpts you attached with your email contain the same data (based on columns 1-3). I suspect that wasn’t your intended use of LARVA. Could you please send me the actual set of annotations you’re using? It would be a huge help to uncovering the root cause of the error.
Q2:
As you said, I think maybe the input annotation file is the point that makes an error. Actually, I do not fully apprehend what the annotation file should be.
In your paper published in 2015, the abstract says: "We make LARVA available as a software tool and release our highly mutated annotations as an online resource (larva.gersteinlab.org).”
So, using the highly mutated annotations you provided may be appropriate. However, this website(“larva.gersteinlab.org”) can not be visited any more. I hope you can provide some help.
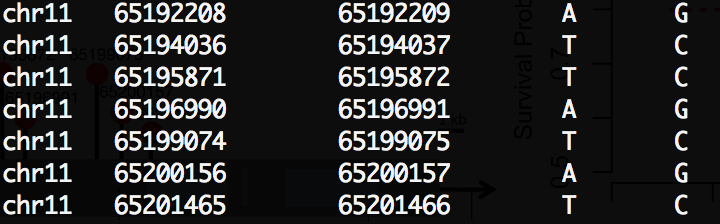
Sorry to bother you for this little things. I used the RegulomeDB annotation file as the LRVAR’s input annotaion file, and the first error I sent you last time was disappeared, but there was a new error like this:
$ processing chromosomes………………….
Error: Invalid length of 0 in annotation file, line 2
Length must be greater than zero
RegulomeDB annotation file(only the first 4 columns were used): [[see image]]
A2:
I apologize for the accessibility issues with the LARVA website. There was a recent change on the backend that messed up the IP address routing to the website. I’ve contacted our IT people about the issue, but until they fix things on their end, the LARVA website can be accessed with its raw IP address: http://54.164.95.124/
Also, concerning your RegulomeDB issue, the reason you get an "Invalid length of 0" error is because the annotation on the second line uses the same coordinate for start and end. The program considers the annotation length to be (end-start), so the second annotation appears to have zero length, which doesn’t really make sense. In fact, it looks like the entire file is made of single nucleotides. This would make sense for the variant file, but for the annotation file, the intention is that the annotations represent intervals on the genome that perform some function. These are typically regions like exons, promoters, enhancers, etc. The idea is to see if these annotations are being hit with a large number of mutations. Single nucleotides don’t really match that annotation definition.
I hope this helps.
-

-
409cc9fd ba26 4343 8e35 cb9869e075e7 image
-
-
filefromclipboard 3.jpg1